Diagnostic des amyotrophies spinales
Les amyotrophies spinales
ce sous-groupe se spécialise dans l'amyotrophie spinale infantile (ASI ou en anglais Spinal Muscular Atrophy (SMA), ou encore amyotrophie spinale proximale) . C'est une maladie neurodégénérative d’origine génétique, de transmission autosomique récessive.

Avec une incidence de 1/6000 soit environ 120 nouveaux cas par an en France, l’amyotrophie spinale infantile constitue la deuxième maladie de transmission autosomique récessive létale de l’enfant la plus fréquente après la mucoviscidose. La fréquence des hétérozygotes dans la population générale est calculée à 1/40. L’ASI se caractérise par une dégénérescence des motoneurones alpha de la corne antérieure de la moelle épinière conduisant à une paralysie progressive touchant la partie proximale des membres et conduisant à une atrophie musculaire. Les patients sont habituellement classés en cinq sous-types cliniques (ASI de type 0 à IV) en fonction de l’âge de début et de la dernière acquisition motrice. Cependant, il est maintenant évident qu’il existe un véritable continuum entre les sous-types définis.
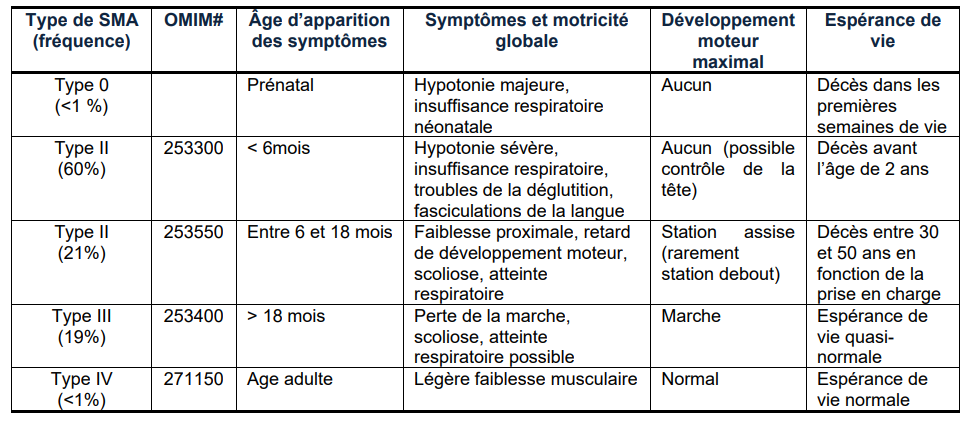
Tableau : Classification des formes et histoire naturelle de la maladie en l’absence de traitement
Source: HAS juin 2023
Le diagnostic est avant tout clinique
Diagnostique clinique : atteinte motrice périphérique, se traduisant par une hypotonie ou une faiblesse musculaire qui prédomine à la racine des membres. Les réflexes ostéo-tendineux (ROT) sont abolis ou diminués.
En 1995, il a été démontré que 95% des patients atteints d’ASI présentent une délétion à l’état homozygote du gène SMN1 (Survival of Motor Neuron). Le gène SMN1 est localisé dans une région de 500 Kb qui s’est dupliquée et inversée au cours de l’évolution. Le gène SMN1, situé dans la région télomérique, possède ainsi un gène copie dans la région centromérique, le gène SMN2, partiellement fonctionnel. Le gène SMN1 code pour la protéine SMN. L’ASI résulte d’une inactivation bi-allélique du gène SMN1. La sévérité de l’ASI est corrélée au taux de protéine SMN fonctionnelle produite, les formes les plus sévères étant associées à de faibles taux de protéine SMN. La protéine SMN est essentiellement produite par le gène SMN1 mais également en faible proportion par le gène SMN2. L’absence totale de copies des gènes SMN1 et SMN2 n’est pas viable. Les patients présentent toujours au moins une copie du gène SMN2 à partir de laquelle une petite quantité de transcrits est générée. Les différences nucléotidiques entre les gènes SMN1 et SMN2 sont mises à profit pour distinguer SMN1 et SMN2 pour le diagnostic moléculaire de la SMA. La délétion à l’état homozygote de l’exon 7 du gène SMN1 conduit au diagnostic de l’ASI dans 95% des cas. Dans 5% des cas, un variant ponctuel intragénique sera associé à une délétion ou une conversion génique du second allèle.
Il existe une corrélation statistique inverse entre le nombre de copies du gène SMN2 et la sévérité du phénotype : 80% des patients atteints d’ASI de type I ont 1 ou 2 copies du gènes SMN2, 82% des patients atteints d’ASI de type II présentent 3 copies du gène SMN2, et 96% des patients atteints d’ASI de type III ont 3 ou 4 copies du gène SMN2. Le gène SMN2 constitue donc le principal gène modificateur de l’ASI par sa capacité à produire une faible quantité de protéine SMN fonctionnelle.
L’élucidation des bases génétiques de l’ASI et de sa physiopathologie a conduit à deux stratégies thérapeutiques innovantes :
- La thérapie génique : Onasemnogene abeparvovec (Zolgensma) commercialisé par la société Novartis.
- La modulation de l’épissage de l’exon 7 du gène SMN1 : Nusinersen (Spinraza) commercialisé par la société Biogen et Risdiplam (Evrysdi) commercialisé par la société Roche.
Déploiement du dépistage néonatal de l’ASI en France
Ces avancées thérapeutiques majeures ont considérablement modifié l’évolution naturelle de la maladie. L’efficacité est d’autant plus marquée que le traitement est initié précocement. Il apparaît donc essentiel de diagnostiquer et traiter le plus tôt possible l’ASI, idéalement à un stade pré symptomatique. Cela a conduit la Haute Autorité de Santé à recommander du déploiement du dépistage néonatal de l’ASI en France.
Pour cette maladie de transmission autosomique récessive, le risque de récurrence est de ¼ à chaque grossesse justifiant une consultation de génétique. Compte tenu de la fréquence de la délétion à l’état hétérozygote du gène SMN1 dans la population générale, une consultation de conseil génétique est indiquée chez les apparentés d’une personne atteinte d’ASI. L’objectif est de préciser le risque qu’ont ces apparentés d’avoir un enfant atteint d’ASI.
En pratique, pour le diagnostic prénatal, une biopsie de trophoblaste est réalisée entre 11 et 13 semaines d’aménorrhée. Le diagnostic direct consistant à rechercher la délétion à l’état homozygote du gène SMN1 est couplée au diagnostic indirect qui explore la ségrégation des allèles morbides dans la famille au moyen de marqueurs microsatellites. Parallèlement, l’analyse des marqueurs microsatellites permet d’exclure toute contamination maternelle du prélèvement fœtal. Pour un diagnostic préimplantatoire, le contact est pris avec l’un des 5 centres agréés pour le diagnostic préimplantatoire (Paris, Strasbourg, Montpellier, Nantes et Grenoble).



