Focus : premiers cas de myopathie mitochondriale par déficit en TK2 (Thymidine kinase) traités par nucléosides en France
Mise à jour lundi 23/03/2026
Actualité
Le déficit en thymidine kinase 2 (TK2d) est une maladie génétique rare, appartenant au groupe des myopathies mitochondriales, causée par des variants pathogènes dans le gène TK2. Cette enzyme joue un rôle essentiel dans
la maintenance de l’ADN mitochondrial, et son dysfonctionnement provoque un syndrome ultra-rare de déplétion et
de délétions multiples de l’ADN mitochondrial entraînant des symptômes variés, allant de formes sévères précoces à des présentations plus tardives et progressives chez l’adulte.
Un traitement expérimental par nucléosides, fondé sur la compréhension des manifestations cliniques et
des mécanismes physiopathologiques du TK2d a permis en 2010 le traitement réussi d’un modèle murin mimant
la maladie TK2d par thérapie nucléotidique.
Un premier essai compassionnel utilisant un traitement par nucléosides (sans effets secondaires graves en dehors
des troubles digestifs) a déjà permis de montrer des résultats très prometteurs.
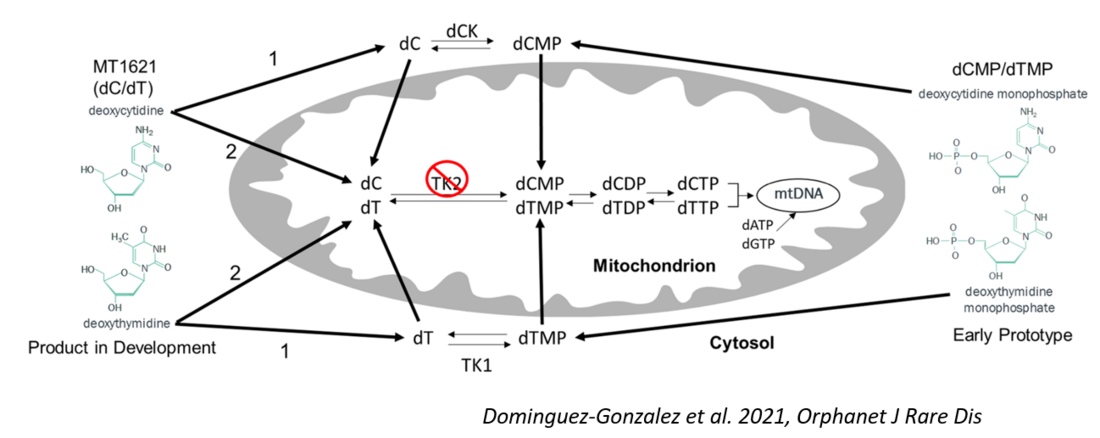
Figure 1 : Mécanisme pharmacologique de la thérapie par nucléosides dans la myopathie associée à TK2d.
Les désoxynucléosides MT1621 (dC/dT) stimulent la production de dTMP et de dCMP respectivement à partir
des enzymes thymidine kinase 1 (TK1) et désoxycytidine kinase (dCK). Ces composés traversent ensuite la membrane mitochondriale afin de fournir les substrats nécessaires à la production de dTTP et de désoxycytidine triphosphate (dCTP) lors de la synthèse et de la réplication de l’ADN mitochondrial (ADNmt). Ces deux mécanismes contribuent à restaurer le pool disponible d’ADN mitochondrial pour la phosphorylation oxydative et la production d’énergie.
L’agence européenne du médicament (EMA) s’est récemment prononcée favorablement début 2026 pour
une autorisation de mise sur le marché (AMM) du Kygevvi® combinant doxecitine et doxribtimine pour les malades atteints de déficit en thymidine kinase 2 (TK2d) avec une symptomatologie survenue avant l’âge de 12 ans.
Afin d’anticiper l’autorisation d’accès précoce à cette thérapeutique, et avoir une meilleure estimation du nombre de patients potentiellement concernés en France la filière FILNEMUS a mené un projet de recensement exhaustif
des patients atteints de déficit en TK2 en France (projet faisant l’objet de la thèse de médecine de Maxime Becker, supervisé par Cécile Rouzier et Vincent Procaccio).
L’objectif principal est d’établir une base de données nationale complète incluant le nombre de patients diagnostiqués, les types de mutations identifiées, ainsi que les différentes formes cliniques observées. Ce travail permettra de mieux comprendre l’épidémiologie française de la maladie, et pouvoir solliciter les cliniciens impliqués de façon réactive lorsque l’accès précoce à cette thérapeutique sera définitivement autorisé en France. Cette étude sera également mise en perspective avec les différentes études internationales déjà réalisées sur les pathologies liées au gène TK2.
Les données de cette cohorte française seront rédigées sous forme d’un article scientifique.